
The Kaderali Lab
Institute of Bioinformatics · University Medicine Greifswald · University of Greifswald
Menu:
Artificial Intelligence and Machine Learning in Medicine
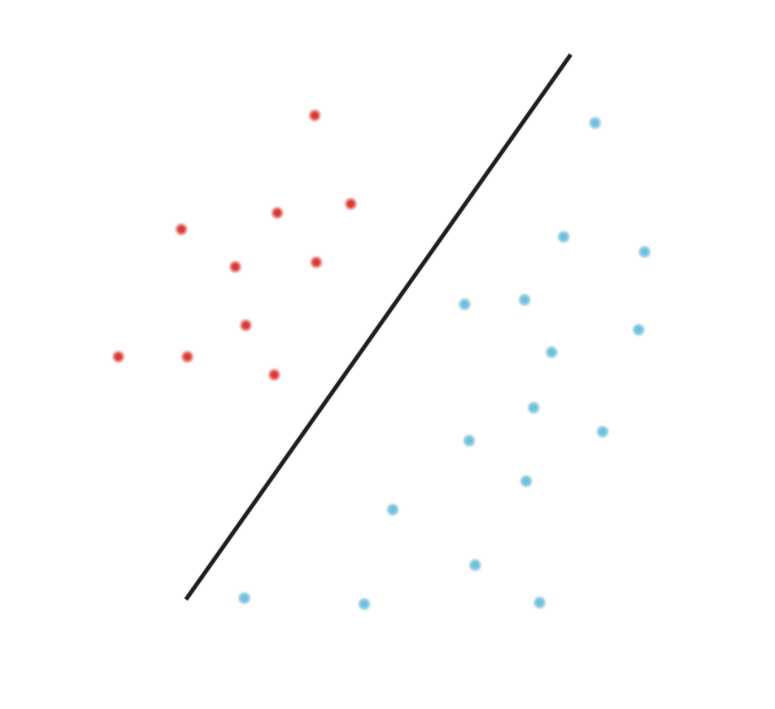
Methods from the Artificial Intelligence field are currently transforming the life sciences. Methods such as Deep Learning, Decision Trees, Support Vector Machines and other AI and machine learning tools transform not only how we analyze data and carry out research, but actually affect how we diagnose disease and treat patients. These methods will transform medicine fundamentally, for patients, medical professionals and researchers alike.
We work at the forefront of
applying artificial intelligence in medicine, from method development
and basic research to translational research at the patient bedside.
Personalized Medicine: Predictive Patterns in Diagnosis and Treatment
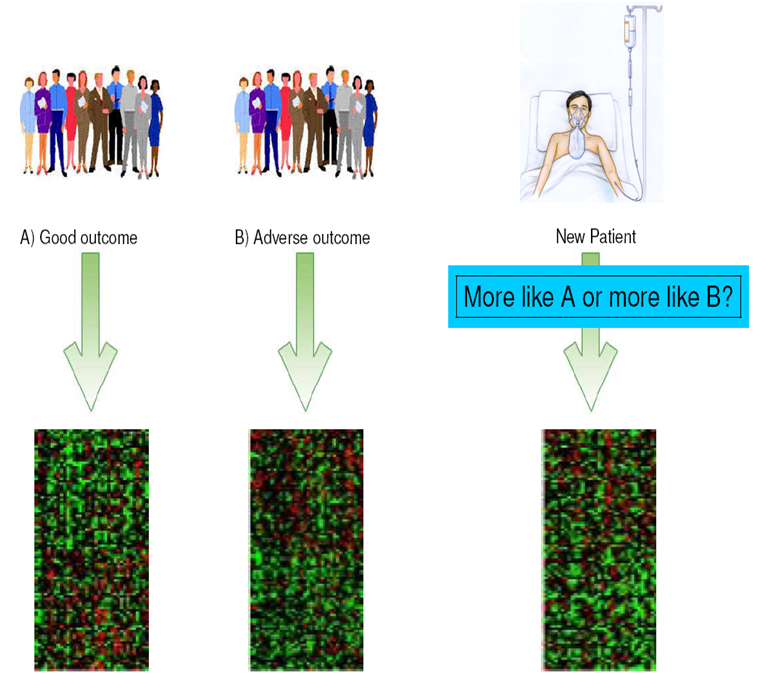
Our behavior and lifestyle, our genes and molecular makeup, and external influences such as exposure to toxic substances and pathogens jointly affect our health.
Due to the complexity of these interactions, it is difficult to predict future health, disease or disease outcome for individual patients. For example, can we predict who will die from cancer, and who from cardiovascular disease? Can we predict how a patient will respond to treatment
with a given drug, whether he or she will experience side effects, and
whether the chosen drug will cure disease? And, having this knowledge:
Can we predict how to best intervene, to change the undesired outcome?
We address questions like this using computer models and artificial
intelligence. By looking at a large number of patients and outcomes,
the computer is able to find patterns that we as humans are unable to
see, and learn over time to make diagnostic and therapeutic decisions,
to support the human doctor.
- Identify genes and pathways that underly disease, to better understand the ethiology of the trait under consideration.
- Identify predictive patterns, that can be used for diagnosis, but also to predict how a patient will respond to a particular treatment, or how the disease will develop in the future.
- Make personalized treatment recommendations, to choose from a set of available drugs the one that will be most efficient in a particular patient.
Network Inference
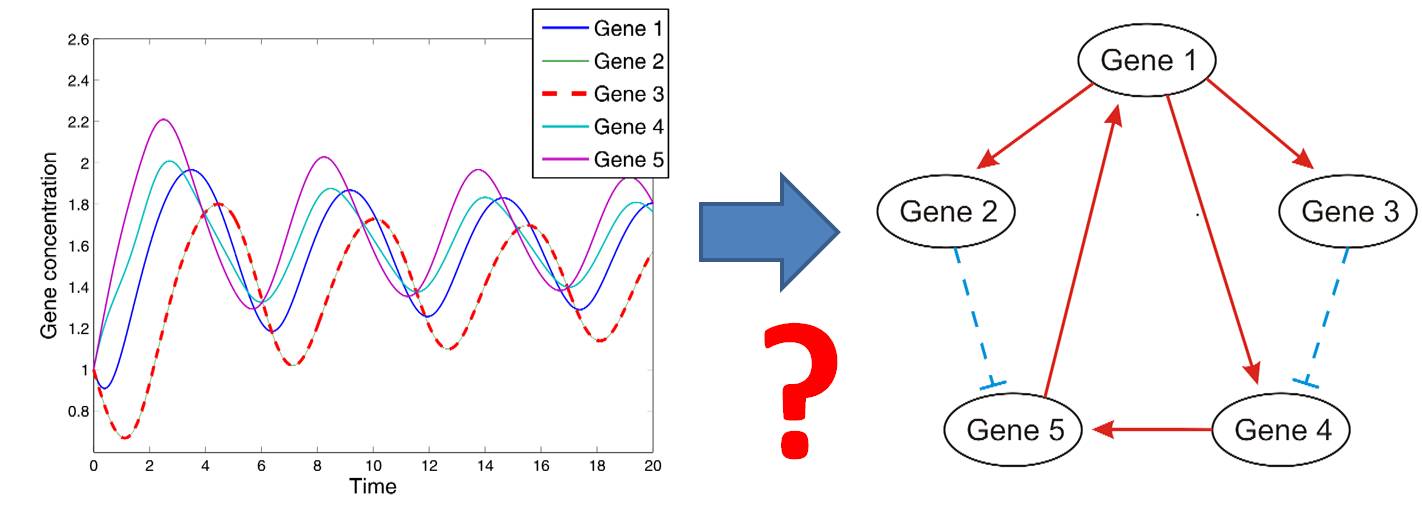
Network inference deals with the problem of reconstructing causal molecular networks e.g. of disease from observations of the networks behavior. Hence, based on mere observational data, for example simply over time, or after certain interventions, the question is to infer how this systems functions internally. This is a complicated inverse problem, that has attracted much attention in engineering ("system identification"), and has tremendous potential for applications in Biology. We work at the forefront of the development of new methods for network reconstrucion in biological applications, using, for example, Bayesian models or nonlinear dynamic systems.